Aladinsane did for 7 years few single crystal neutron diffraction experiments, and lenghty analyses (magnetic structure 2006-2010 and structure 2011-2013) almost completely alone. As well summarised in the song "Orient Express" says, "[his] mystery is in your own...": you figure it out!... surrounded by "nasty [Diamond] DOGS" never ready to aknowledge how difficult it was to get clean results, how much this is due to the fact the analyses are
becoming incredibly harder at x=0.5 compared to x=0.4 (Aladinsane's original thesis
work) because of the larger deviation of the orthorombic cell
parameters from an avergage cubic perovskite, which is splitting
more the twin
contributions in reciprocal space and make the whole data analysis a
real (but worthwhile) nighmarre to fight against.
The magnetic structure part
of this work, (I recall, Aladinsane spent 4 years of analyses 2006-2010 on that part...) rather trivially obtains the known
CE-type order and even if it is a single crystal work, twinning and high model degeneracy imposes to use highly constrained models (3 to 5 parameters max). This represent a huge work not even guaranteed to be complete/discriminative in terms of possible magnetic domains superimposed to twin domains which makes the sought for non-collinear magnetic order still candidate!
The (so far privileged) collinear structure result, verifying a
structure known since the 50's is therfore in that respect quite
unfairly not getting so much new insights though it is (from the
exchange topology point of view, as discusssed in the linked paper) an
extrermely important check to be done....
Severe
technical
difficulties in the final most promising part of the work, i.e in the study, later recentred on the atomic structure determination after 2011 (e.ven if it is simpler, Daoud-Aladine still faced unforseen difficulties here, i.e. unstable refinements requiring more data clean up and/or software debugging, impossible to deal with without HELP!), have not been sorted out before... so the result never reached crystallographic standards giving precise
refinements that could enable a highly desirable comparison between Pr0.6Ca0.4MnO3 and Pr1/2Ca1/2MnO3. This is the only reason why that has not been published, though independently, regarding the physical interpretation, even in its preliminary state the structural analysis provides an unambiguous favor for ZP ordering
(bond centred) and related symmetry
and justify the idea that the original structure which is an
approximation of the real superstructure of x=0.4, provides a good approximation also for Pr1/2Ca1/2MnO3.
I'm hence retrospectively making clear once for the good, why such structural analyses provide approximate or average strucutres, but also why the "approximations" are of different nature in the two analysis and why they are fully justified/valid and still constitute the best structural model to date. In Pr0.6Ca0.4MnO3, Aladinsane refined an averaged structure on a subset of the superstructure
reflections found in the ground state. The compound's off composition from x=0.5 may manifest indeed
the development at lower temperatures (T<~90K) of an extra set of
superstructures (i.e an extra distortion, seen at x=0.375, but that can be short range nearby), that the superstructure
model determined in Pr0.6Ca0.4MnO3 at a temperature far above (T=190K) thermally averages
out. This is why in his original work, where he have determined the model of the HT charge
ordered phase containing only the main subset of supprstructurwe lines (with excellent bragg factors by the way), he could claim that it provides a natural averaged model for Pr1/2Ca1/2MnO3, which shows the same subset of superstructure line analysed in Pr0.6Ca0.4MnO3, as a unique set.
What was an "approximation & hypothesis for interpretation", is now proven to be essentially valid by what's so far, a semi-qualitative check (at T=190K) in the real Pr1/2Ca1/2MnO3. This is done "at first approximation"
also, but the term "approximation" here, has a different meaning: it is
due to the fact the analysis is not complete, and consist of fixing a structure - the "approximate/averge" one determined in Pr0.6Ca0.4MnO3 as described above,
which give in a first step worse bragg factors. As mentionned above,
this is because the more severe twining makes the analysis so tricky, that the true structure could not be fully refined. Still, regarding physical intepretations, the check is good enough to discriminate the competing symmetries unambiguously.
Still, continuing with semi-qualitave arguments at x=0.5 he additionally measured the temperature dependence of selected superstructure lines theoretically sensitive to eventual changes of of symetry
back to a site centred charge order. This is a new information he had not looked for in at x=0.4
because it is actually easier to follow it at x=0.5, where luckily, structural superlattice reflections (q=(0 1/2 0)) are not polluted by magnetic contributions of other
twins domains (with k=(1/2 0 0)) (see all the figures below): this, additonal information confirms that the "approximate" structure with the symmetry interpreted as ZP order, do not change down to 10K.
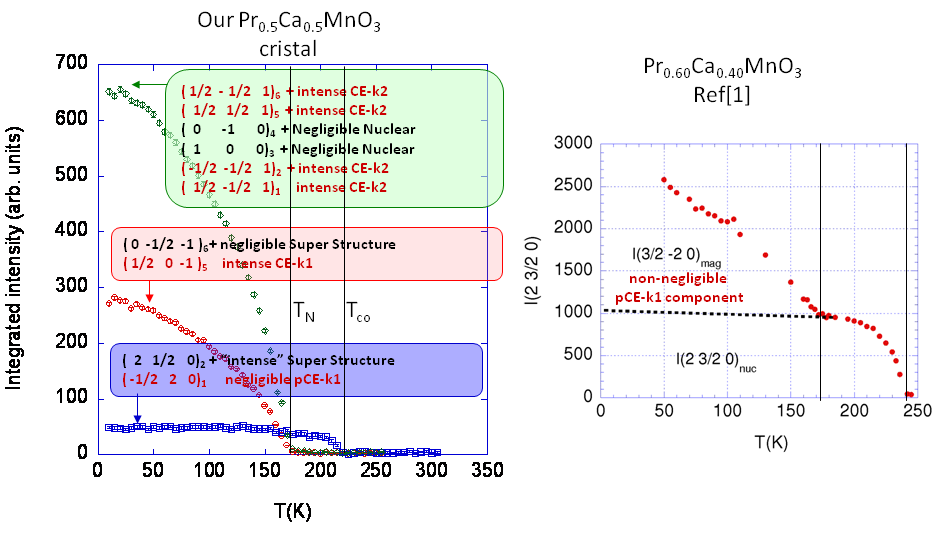
How twinnin mixes twin domain contribution is illustrated above (left figure). Note the abscence of intensity changes of the superstructure peaks (blue square) at T_N in this very
1st measurements proving that the sample is a pure half-doped PCMO, in
comparison to the sensitivity of similar peaks to magnetic ordering due
to contribution from other twins, in Pr0.6Ca0.4MnO3 (right figure). This behaviour of superstructure peaks
has been more systematically investigated in the last experiment on Pr1/2Ca1/2MnO3 (see
below) on appropriately selected reflections like the one that is
circled out below: this is what led me conclude to K.R on face book "C'est bon, il n'y a pas de transition à TN". I have no back up, but the temperature dependence of
the integrated intensity of such peaks is very easy to retrieve, and
all show a behaviour similar to the "blue" peak above, i.e have no
accident at TN).
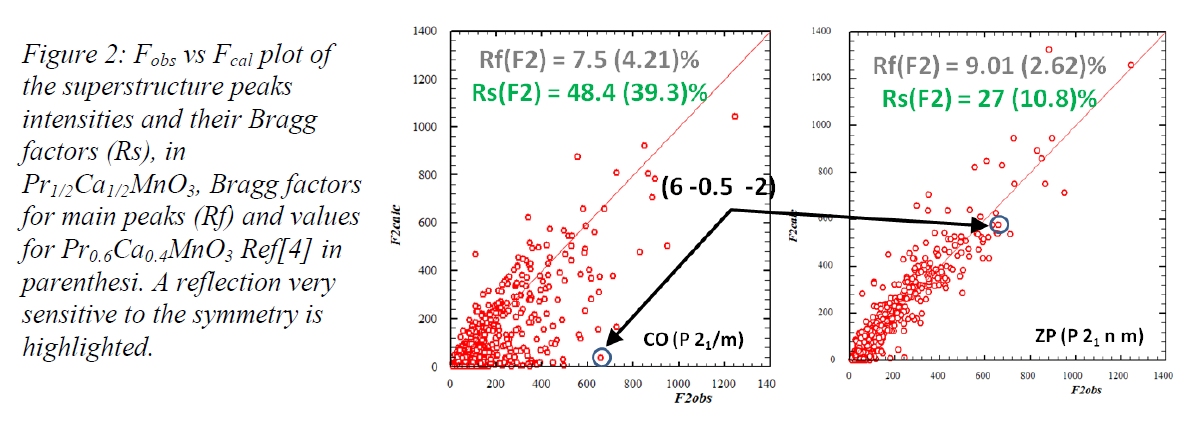
The calculation and bragg
factors shown above are taken from a proposal showing the RESULT of the ongoing analysis
of the
1st experiment dedicated to the structure determination at T=190K,
still restricted
to date to fixed models. This check (T=190K) therefore show that since
October 2012 (proposal submission date), the published structure of Pr0.6Ca0.4MnO3 was known to fit data on Pr1/2Ca1/2MnO3
at "first approximation", and
the experiment done in march 2013,
proves
(see main text above) that
PCMO50 has P 21 n m Symmetry
Down to 10K